

在前期的介绍中,小编分别为大家介绍了分子对接的案例以及适用范围,看完之后是不是跃跃欲试呢?那么,在本期中,小编将为大家介绍一款常用分子对接软件的使用!
分子对接(molecular docking)依据配体与受体作用的“锁-钥原理”(lock and key principle),模拟小分子配体与受体生物大分子相互作用。配体与受体相互作用是分子识别的过程,主要包括静电作用、氢键作用、疏水作用、范德华作用等。通过计算,可以预测两者间的结合模式和亲和力,从而进行药物的虚拟筛选。
考虑到大家为首次接触分子对接软件的操作,因此本教程选取了操作十分便捷的分子对接软件——LeDock1,并以神经氨酸苷酶(neuraminidase)为例,来介绍其使用。选取的蛋白配体复合物晶体结构文件为3cl0,可从PDB数据库上下载:http://www.rcsb.org,亦可在我们分享的网盘中获取。
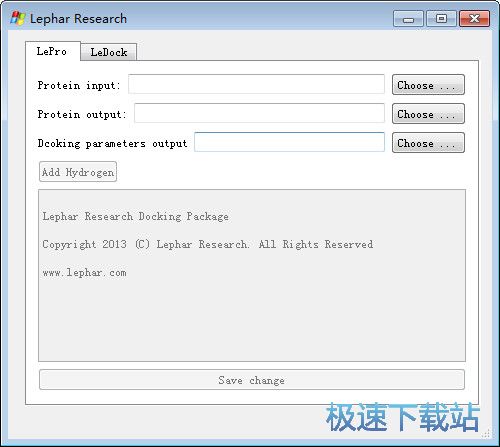
软件界面
LeDock共含有2个标签,分别为LePro与LeDock。其中,LePro用于蛋白的处理(如删去蛋白中的水分子、离子、金属、配体、辅因子等),并会生成用于下一步分子对接的in文件,而LeDock则是在各项参数确定了之后进行分子对接。
软件操作
一、蛋白准备
在LePro标签的Protein input中选择3cl0.pdb的路径(注意路径中不能含有中文),然后软件会自动在选择在该路径下生成pro.pdb(处理过后的蛋白的pdb文件)及dock.in(用于下步分子对接的参数文件),点击Add Hydrogen,上述2个文件就会立马生成。
dock.in文件包括了均方根偏差(RMSD),Binding pocket及Number of binding poses。RMSD表示对接出来的构象聚类时RMSD的截断值;Binding pocket表示结合位点的3维坐标,分别为xmin xmax,yminymax,zmin zmax;Numberof binding poses表示生成的构象个数,经聚类后产生的构象数会少于之前所生成的数目。
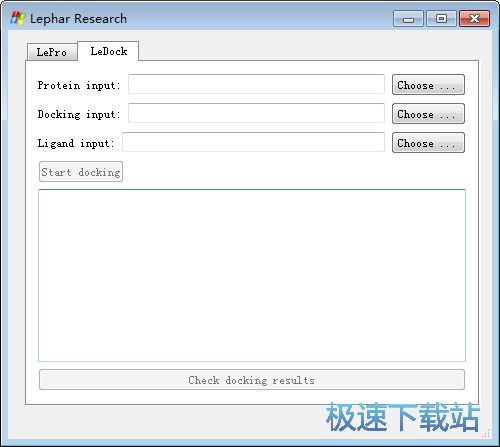
二、分子对接
在LeDock标签的Protein input,Docking input中分别指定上一步生成的pro.pdb与dock.in文件的路径,在Ligand input中指定需要进行对接的配体——lig.mol2的路径(该路径需和前两个文件的路径相同,且对接的分子需要为mol2格式)。比较后,点击Start docking,会生成lig.dok文件(对接产生的化合物构象所在文件),该文件可用Pymol来观察化合物的构象。
三、Pymol的使用
打开Pymol,选择菜单栏中的File>Open...,在文件类型中选择ALL Files,再找到lig.dok的文件,双击打开,即可查看生成的化合物的构象。
若要查看其与蛋白结合的情况,可再按上述方法打开pro.pdb文件,再在图形界面的pro栏中点击S>as>cartoon,即可以cartoon的模式来显示蛋白以得到更为清楚的图像。
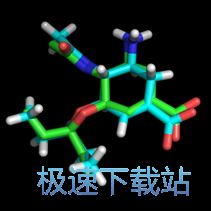
若要比较对接的精度,可以打开3CL0.pdb文件,该晶体文件中本身便有配体,而我们也正是使用该配体进行重对接,来评估对接的精度,在3CL0栏中点击A>preset>ligand sites>cartoon,即可查看到晶体中的配体与蛋白的相互作用模式,再将生成的构象与其比较即可判断对接的精度。(注意:生成的构象为lines的显示模式,且含有氢,而晶体结构中的配体不含氢,且此时其为sticks模式,为更好地观察,可进行下述操作:分别在生成的构象对应的栏中选择S>as>sticks,再在3CL0栏中选择A>hydrogens>add即可)。图2即为晶体结构中的配体构象与对接后打分比较高的配体构象,两者是不是非常接近呢!
文件信息
文件大小:701952 字节
MD5:407A739622A40984673F5B6BCD4D1735
SHA1:F57C029D0BD761A7FD3FECE9A6E68087D6D1AFA9
CRC32:53CCE558
- 共 0 条评论,平均 0 分 我来说两句
- 人气英雄联盟下载官方下载
- 《英雄联盟》(简称LOL)是由美国拳头游戏(Riot Games)开发、中国大陆地区腾讯游戏代理运营的英雄对战MOBA... [立即下载]
- 好评微信电脑版 2.6.6.44 官方版
- 在微信越来越普及的时代下,微信的用户量是一天比一天高。但是微信有一个缺点,它不像QQ那样可以方便地在电... [立即下载]